|
Schedule: Fee: $500 per learner Agenda: CEUs: Demonstration: |
Adverse Event & Effect Workshop:
Adverse event reporting is one of the more difficult aspects of conducting a medical device clinical trial. The rules are different from drugs and different in the States and Europe.
Even the definition of what is and isn't an adverse event can vary. Agenda: Description and Learning
Objectives: Learning Objective: Course level: You will receive: Who should attend: 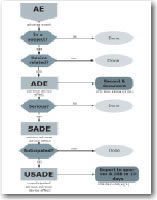
Presentation—3 hours
Quiz—1 hour
Course Assessment—10 minutes
In this workshop you'll be taken step-by-step through the definitions of the different types of adverse events and review the recording and reporting requirements for both the US and EU. You'll learn a logical flow-chart plan to what needs to be done, so that both monitors and investigators can follow along. You'll learn what it means for a sponsor to "evaluate" an adverse event. You'll look at warning letters to IRBs, investigators, and sponsors to see how FDA views adverse events. You'll look at hypotheticals and decide what to do. Approaching the learning task using many different tools, we'll use lecture, slides, colorful flowcharts, decision-making polls, and a challenging quiz to master this well-researched material.
The goal of this workshop is to learn a decision-making framework that will facilitate correct adverse event recording and reporting.
This is an intermediate level course. Participants are expected to have a
basic knowledge of clinical research and device regulations.
1) One computer connection for one learner.
2) Printable copy of PowerPoint slides.
3) Adverse event & Effect flowcharts.
4) A searchable pdf file of 2005 CDRH warning letters.
5) Online exam with immediate score results.
6) Certificate of Attendance and 0.4 CEUs.
[x] Clinical research professionals.
[x] Regulatory professionals.
[x] Managers and decision-makers.
[x] Executives for start-up companies.
[x] Investigators for medical device trials.
[x] DSMB and CEC members.
[x] IRBs who review medical device trials.